





Окрім технології, синтез глікозидів завжди був цікавим для науки, оскільки це дуже поширена реакція в природі. Нещодавні статті Шмідта, Тосіми та Тацути, а також численні посилання, цитовані в них, прокоментували широкий спектр синтетичних можливостей.
У синтезі глікозидів багатоцукрові компоненти поєднуються з нуклеофілами, такими як спирти, вуглеводи або білки. Якщо потрібна селективна реакція з однією з гідроксильних груп вуглеводу, всі інші функції повинні бути захищені на першому етапі. В принципі, ферментативні або мікробні процеси, завдяки своїй селективності, можуть замінити складні етапи хімічного захисту та депротекції для селективного видалення глікозидів з ділянок. Однак, через довгу історію алкілглікозидів, застосування ферментів у синтезі глікозидів не отримало широкого вивчення та застосування.
Через можливості відповідних ферментних систем та високі виробничі витрати, ферментативний синтез алкілполіглікозидів не готовий до модернізації до промислового рівня, тому перевагу надають хімічним методам.
У 1870 році М.А. Коллі повідомив про синтез «ацетохлоргідрози» (1, рисунок 2) шляхом реакції декстрози (глюкози) з ацетилхлоридом, що зрештою призвело до історії шляхів синтезу глікозидів.
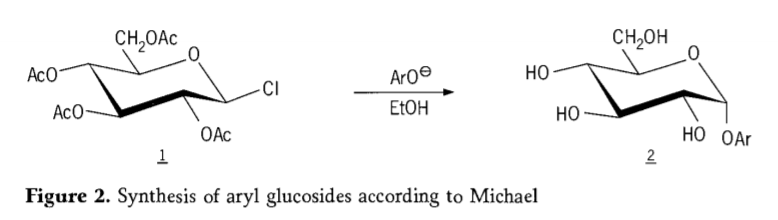
Тетра-0-ацетил-глюкопіранозилгалогеніди (ацетогалогоглюкози) пізніше виявилися корисними проміжними продуктами для стереоселективного синтезу чистих алкілглюкозидів. У 1879 році Артур Майкл успішно отримав чіткі, кристалізовані арилглікозиди з проміжних продуктів та фенолатів Коллі (Аро-, рисунок 2).
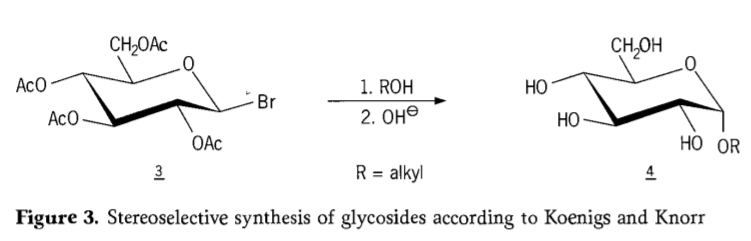
У 1901 році Майкл здійснив синтез широкого спектру вуглеводів та гідроксильних агліконів, коли В. Кенігс та Е. Кнорр запропонували свій удосконалений стереоселективний процес глікозидування (Рис. 3). Реакція включає заміщення SN2 біля аномерного вуглецю та протікає стереоселективно з інверсією конфігурації, утворюючи, наприклад, α-глюкозид 4 з β-аномеру проміжного продукту ацеобромглюкози 3. Синтез Кенігса-Кнорра відбувається у присутності срібних або ртутних промоторів.
У 1893 році Еміль Фішер запропонував принципово інший підхід до синтезу алкілглюкозидів. Цей процес зараз добре відомий як «глікозидування Фішера» та являє собою кислотно-каталізовану реакцію глікоз зі спиртами. Проте будь-який історичний звіт повинен також включати першу зареєстровану спробу А. Готьє в 1874 році перетворити декстрозу безводним етанолом у присутності хлоридної кислоти. Через помилковий елементний аналіз Готьє вважав, що отримав «диглюкозу». Пізніше Фішер довів, що «диглюкоза» Готьє насправді була переважно етилглюкозидом (рис. 4).
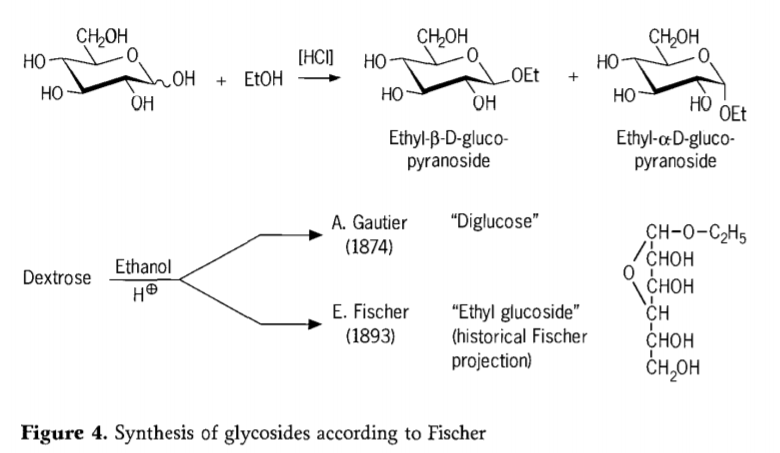
Фішер правильно визначив структуру етилглюкозиду, що видно із запропонованої історичної фуранозидної формули. Фактично, продукти глікозидування Фішера є складними, здебільшого рівноважними сумішами α/β-аномерів та ізомерів піранозиду/фуранозиду, які також містять випадково зв'язані глікозидні олігомери.
Відповідно, окремі молекулярні види нелегко виділити з реакційних сумішей Фішера, що було серйозною проблемою в минулому. Після деякого вдосконалення цього методу синтезу Фішер згодом застосував синтез Кенігса-Кнорра для своїх досліджень. Використовуючи цей процес, Е. Фішер та Б. Гельферіх першими повідомили про синтез довголанцюгового алкілглюкозиду, що проявляє властивості поверхнево-активної речовини, у 1911 році.
Ще в 1893 році Фішер правильно помітив важливі властивості алкілглікозидів, такі як їх висока стійкість до окислення та гідролізу, особливо в сильнолужних середовищах. Обидві характеристики є цінними для алкілполіглікозидів, що використовуються як поверхнево-активні речовини.
Дослідження, пов'язані з реакцією глікозидування, все ще тривають, і нещодавно було розроблено кілька цікавих шляхів отримання глікозидів. Деякі процедури синтезу глікозидів наведено на рисунку 5.
Загалом, процеси хімічного глікозидування можна розділити на процеси, що призводять до складних олігомерних рівноваг у кислотно-каталізованому глікозильному обміні.
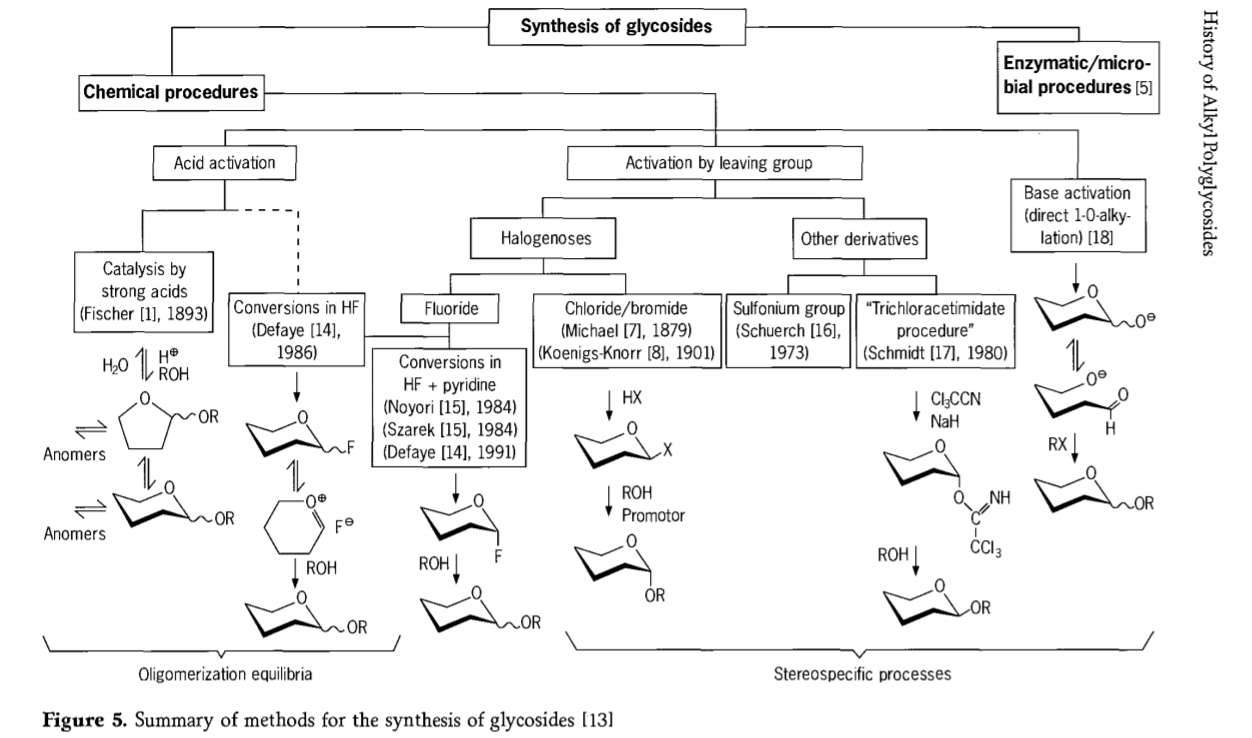
Реакції на відповідно активованих вуглеводних субстратах (глікозидні реакції Фішера та реакції фтористого водню (HF) із незахищеними молекулами вуглеводів) та кінетично контрольовані, незворотні та переважно стереотаксичні реакції заміщення. Другий тип процедури може призвести до утворення окремих видів, а не до складних сумішей реакцій, особливо у поєднанні з методами консервативних груп. Вуглеводи можуть залишати групи на ектопічному вуглеці, такі як атоми галогенів, сульфоніли або трихлорацетимідатні групи, або активуватися основами перед перетворенням на трифлатні ефіри.
У окремому випадку глікозидавання у фтористому водні або в сумішах фтористого водню та піридину (піридиній полі[фтористий водень]), глікозилфториди утворюються in situ та плавно перетворюються на глікозиди, наприклад, за допомогою спиртів. Було показано, що фтористий водень є сильно активуючим, недеградуючим реакційним середовищем; рівноважна автоконденсація (олігомеризація) спостерігається подібно до процесу Фішера, хоча механізм реакції, ймовірно, відрізняється.
Хімічно чисті алкілглікозиди підходять лише для дуже спеціальних застосувань. Наприклад, алкілглікозиди успішно використовуються в біохімічних дослідженнях для кристалізації мембранних білків, таких як тривимірна кристалізація порину та бактеріородопсину в присутності октил β-D-глюкопіранозиду (подальші експерименти, засновані на цій роботі, призвели до Нобелівської премії з хімії для Дейзенхофера, Губера та Мішеля в 1988 році).
Під час розробки алкілполіглікозидів стереоселективні методи використовувалися в лабораторних масштабах для синтезу різноманітних модельних речовин та вивчення їхніх фізико-хімічних властивостей. Через їхню складність, нестабільність проміжних продуктів, кількість і критичний характер відходів процесу, синтези типу Кенігса-Кнорра та інші методи захисних груп створювали б значні технічні та економічні проблеми. Процеси типу Фішера порівняно менш складні та легші для виконання в комерційних масштабах, і відповідно є кращим методом для виробництва алкілполіглікозидів у великих масштабах.
Час публікації: 12 вересня 2020 р.